







La validación del proceso de esterilización con autoclave es el paso que convierte este procedimiento en un proceso demostrable, repetible y defendible ante auditorías.
No se trata de “hacer pruebas” aisladas, sino de documentar que la esterilización con autoclave cumple su objetivo, de forma consistente, bajo condiciones definidas.
La norma ISO 17665 es la referencia internacional más utilizada para estructurar esta validación en procesos de calor húmedo (vapor).
En ella se establecen los requisitos para el desarrollo, la validación y el control rutinario del proceso.
En el entorno hospitalario, donde las cargas son variadas y la demanda cambia por urgencias quirúrgicas, esta estructura evita un error común: asumir que “si el ciclo corrió, esterilizó”.
A lo largo de esta guía se explican los conceptos, el método IQ/OQ/PQ, los registros, los indicadores y las decisiones técnicas que más pesan en una validación sólida.
Al final tendrás claridad sobre qué es la validación de autoclaves, cuál es el método, cómo verificar rutinas, y cómo alinear la validación con normas colombianas.
Qué es la validación del proceso de esterilización con autoclave y por qué es crítica
En esterilización la palabra “validación” significa una cosa muy concreta: evidencia documentada de que el proceso, aplicado a cargas definidas, logra resultados consistentes dentro de límites preestablecidos.
La norma ISO 17665 formaliza esa idea al exigir que el proceso se desarrolle, se valide y luego se controle rutinariamente con criterios definidos.
Dicho de forma operativa, el autoclave de la central de esterilización puede tener pantalla, impresora y alarmas modernas.
Sin embargo, si no existe evidencia de desempeño en cargas representativas, el proceso queda vulnerable ante fallas silenciosas como:
- Aire atrapado
- Vapor de calidad inadecuada
- Errores de carga
- Secado deficiente
- Medición sin trazabilidad.
Validación vs. verificación rutinaria: no son lo mismo
La verificación rutinaria busca confirmar día a día que el proceso sigue “bajo control”.
La validación, en cambio, demuestra que el proceso funciona en condiciones definidas y que los controles rutinarios son adecuados para detectar desviaciones.
Un ejemplo ayuda: la prueba Bowie-Dick (cuando aplica) evalúa remoción de aire y penetración de vapor en esterilizadores de pre-vacío.
Sin embargo, esta prueba por sí sola no certifica la esterilidad del instrumental.
En guías técnicas se presenta como prueba de desempeño/función del esterilizador para detectar problemas como aire residual o gases no condensables.

Qué evidencia exige un proceso controlado según la norma ISO 17665
La norma ISO 17665 define el marco: proceso definido, parámetros críticos, criterios de aceptación, validación documentada y control rutinario con registros.
En hospitales, la guía ISO/TS 17665-2 (documento de orientación) reconoce que son comunes las cargas mixtas.
Adicionalmente, que el enfoque práctico debe apoyarse en familias de carga, criterios operativos y buenas prácticas de reprocesamiento.
Normas y marco aplicable en Colombia para validar la esterilización con autoclave
Una validación bien hecha en Colombia se sostiene sobre dos pilares:
- Normativa nacional para reprocesamiento/esterilización en prestadores
- Estándares técnicos internacionales para estructurar la evidencia.
Manual de Buenas Prácticas de Esterilización (Resolución 2183 de 2004)
La Resolución 2183 de 2004 adopta el Manual de Buenas Prácticas de Esterilización para prestadores de servicios de salud.
En consecuencia, lo presenta como la herramienta para unificar criterios y requisitos mínimos en centrales de esterilización.
Aunque el manual no reemplaza los estándares ISO, sí fija una expectativa clara.
Procesos estandarizados, documentación y condiciones mínimas que deben sostener la calidad del reprocesamiento.
Buenas Prácticas de Reprocesamiento DMER (Resolución 914 de 2025) y habilitación (Resolución 3100 de 2019)
La Resolución 914 de 2025 adopta el Manual de Requisitos para la Implementación de Buenas Prácticas de Reprocesamiento de Dispositivos Médicos y Elementos Reutilizables (DMER).
Esta normativa es aplicable tanto a prestadores como a operadores externos de esterilización que presten etapas del proceso.
En paralelo, la Resolución 3100 de 2019 regula procedimientos y condiciones de inscripción/habilitación de servicios de salud.
Adicionalmente cuenta con un anexo técnico que exige condiciones para procesos prioritarios, entre ellos los relacionados con esterilización y reprocesamiento.
Un punto importante: el DMER incluye definiciones y expectativas de seguridad.
Por ejemplo, se usa el concepto de garantía de esterilidad (típicamente 10⁻⁶) como referencia para dispositivos médicos.
Lo anterior refuerza la necesidad de procesos controlados y evidenciables.
Cómo se conectan ISO 17665 / EN 285 con el contexto colombiano
En la práctica, la norma ISO 17665 ayuda a responder “cómo demostrar” el control del proceso.
Por otra parte, EN 285 (Norma Europea de Esterilización) se usa como referencia técnica frecuente en esterilizadores de vapor grandes.
Se incluyen además pruebas y consideraciones sobre penetración de vapor y fallos por aire, fugas o gases no condensables.
Así, validar “según normas colombianas” suele significar cumplir el marco nacional (DMER/Manual).
Sin embargo, también se debe estructurar la evidencia con un estándar reconocido internacionalmente (ISO 17665), dejando trazabilidad documental y técnica consistente.
Definir el proceso antes de validar: alcance, cargas, parámetros y criterios de aceptación
Antes de correr sensores, hay una regla que evita la mayoría de las validaciones frágiles: no se puede validar lo que no está definido.
Definir el proceso incluye el tipo de equipo, el ciclo, las cargas, el empaque, los parámetros críticos y los criterios de aceptación.
Qué se valida exactamente en un sistema de esterilización con autoclave
La validación del proceso de esterilización con autoclave no se limita al “ciclo estándar”. Incluye:
- Equipo de esterilización autoclave (instalación, instrumentos, registradores, alarmas),
- Ciclo (etapas, setpoints, tolerancias),
- Tipo de carga y configuración (familias y peor caso),
- Empaque y el método de preparación,
- Calidad del vapor y condiciones de utilidades.
La guía de la Organización Mundial de la Salud sobre descontaminación y reprocesamiento resalta es puntual con respecto al proceso.
Exige que la calidad del vapor y su generación/distribución deben diseñarse y mantenerse para producir un suministro confiable, sin interferir con el proceso ni dañar los dispositivos.
Selección de familias de carga y “peor caso”
En hospitales no es realista validar “cada set” como si fuera un producto industrial único.
Por eso se trabaja por familias de carga (textiles, instrumental metálico, contenedores rígidos, cargas porosas, dispositivos con lúmenes, etc.)
Adicionalmente, se debe eligir un “peor caso” para cada familia.
La norma ISO/TS 17665-2 reconoce que las cargas mixtas son comunes en entornos de salud.
Adicionalmente, que la validación debe apoyarse en buenas prácticas, configuraciones definidas y criterios prácticos de control.
Un peor caso no es “la carga más pesada” necesariamente. Suele ser la combinación que dificulta más la penetración del vapor o el secado:
- Porosidad alta
- Contenedores densos
- Empaques múltiples
- Lúmenes
- Conjuntos con alta masa térmica en zonas menos expuestas.
Conceptos clave: letalidad, esterilidad y nivel de garantía
Para evitar malentendidos, conviene recordar que “estéril” es un concepto probabilístico en términos de garantía de esterilidad.
En el marco DMER se menciona el nivel 10⁻⁶ como referencia para dispositivos médicos.
La consecuencia práctica es simple: la validación busca establecer que el proceso, bien ejecutado, alcanza la letalidad requerida.
En consecuencia, los controles rutinarios deben ser capaces de alertar si el proceso se aparta de lo validado.
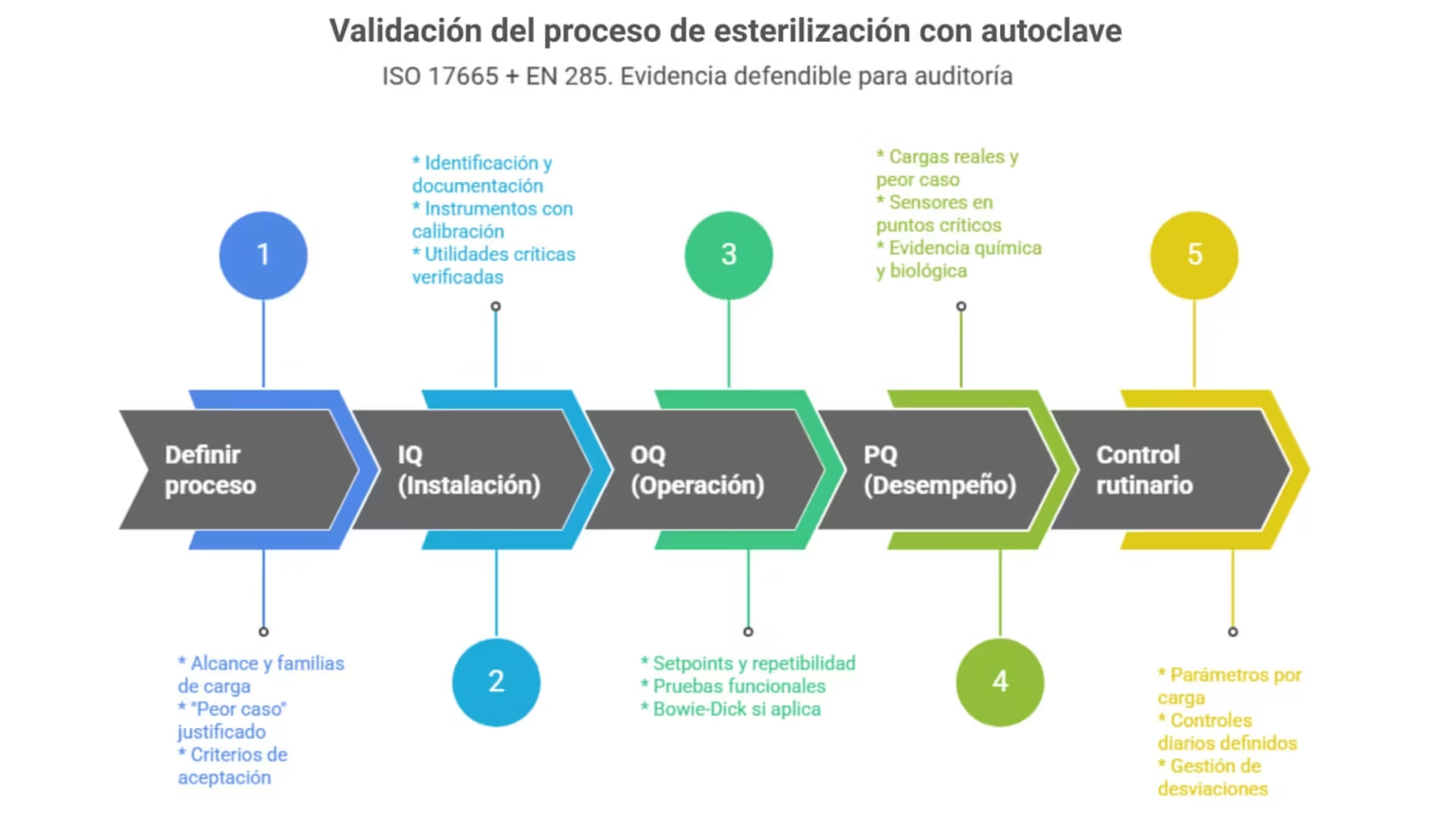
Método de validación del autoclave con enfoque IQ/OQ/PQ
Aquí aparece una de las preguntas más repetidas: ¿cuál es el método de validación del autoclave?
En la mayoría de programas hospitalarios y farmacéuticos se estructura como IQ/OQ/PQ, porque organiza evidencia de forma lógica y auditable.
La norma ISO 17665 exige validación y control rutinario; ISO/TS 17665-2 amplía la orientación práctica.
¿Cuáles son los 3 tipos de validación? (IQ, OQ y PQ)
En el lenguaje cotidiano del sector, los “tres tipos” suelen ser:
- Calificación de instalación (IQ): evidencia de instalación correcta según especificación.
- Calificación de operación (OQ): evidencia de operación correcta en su rango, con pruebas funcionales.
- Calificación de desempeño (PQ): evidencia de desempeño con cargas reales/peor caso.
Esta estructura es compatible con la forma en que ISO 17665 aborda la validación como parte del ciclo de vida del proceso.
Gestión del cambio: cuándo revalidar o recalificar
Un proceso validado puede dejar de estarlo si se cambia algo “relevante”. Algunos disparadores típicos:
- Reemplazo de sensores o componentes críticos,
- Cambios de software/firmware,
- Modificaciones en suministro de vapor/agua,
- Cambio de empaque o contenedor,
- Incorporación de nuevos dispositivos con diferente complejidad,
- Variación sostenida en resultados (por ejemplo, cargas húmedas recurrentes).
ISO/TS 17665-2 enfatiza la buena práctica y el control del proceso a lo largo del tiempo, lo cual se traduce en gestionar cambios y mantener evidencia vigente.
IQ: Calificación de instalación del equipo de esterilización autoclave
La IQ responde una pregunta básica: ¿el equipo quedó instalado de manera que pueda operar como fue diseñado? Aquí se gana o se pierde mucha credibilidad documental.
Requisitos de usuario, documentación del fabricante y trazabilidad
Una IQ sólida incluye, como mínimo:
- Identificación del esterilizador (marca/modelo/serie),
- Manuales y recomendaciones del fabricante,
- Lista de instrumentos que miden (temperatura, presión, tiempo) y su estado de calibración,
- Diagramas de instalación y conexiones,
- Verificación de condiciones de seguridad eléctrica y mecánica.
En este punto, no se trata de “llenar formatos”. Se trata de asegurar que el registro que saldrá del equipo es confiable para defenderlo en auditoría.
Utilidades críticas: vapor, agua, drenajes, electricidad y seguridad
La IQ debe verificar que el suministro de vapor y su calidad no comprometen la esterilización.
La OMS subraya que el sistema de agua, generación de vapor y tuberías deben diseñarse, construirse y mantenerse para entregar vapor de calidad controlada.
En un enfoque práctico, aquí también se documenta la configuración de drenajes, trampas de vapor, pendientes y retornos.
De hecho, problemas de condensado o arrastre pueden afectar el secado y la estabilidad térmica del ciclo.
OQ: Calificación de operación y pruebas funcionales
La OQ busca demostrar que el autoclave opera correctamente en sus rangos y que sus funciones críticas responden como deben.
Es el puente entre “está instalado” y “esteriliza cargas reales”.
Distribución térmica y repetibilidad de control
En OQ se evalúa si el equipo alcanza y mantiene los setpoints, y si la instrumentación responde de manera consistente.
Dependiendo del enfoque del programa, se pueden hacer corridas vacías o con cargas simuladas, siempre con criterios definidos.
El valor de esta fase es detectar inconsistencias antes de entrar a PQ. Si en OQ hay variabilidad alta, cualquier PQ quedará discutible.
Remoción de aire, gases no condensables y prueba Bowie-Dick
En esterilizadores de pre-vacío, la remoción de aire es crítica. La norma EN 285 advierte que fallos en pruebas de penetración de vapor pueden explicarse por remoción de aire ineficiente, fugas o presencia de gases no condensables en el suministro de vapor.
La prueba Bowie-Dick es un estándar de facto para evaluar la eficiencia de remoción de aire en ciclos de pre-vacío.
La norma ISO 11140-5 establece requisitos para indicadores clase 2 usados en pruebas tipo Bowie-Dick para evaluar la efectividad de remoción de aire durante la fase de pre-vacío.
Además, en programas de aseguramiento de calidad se recomienda una rutina diaria.
Un resumen de guías de AAMI ST79 indica que la prueba Bowie-Dick debe realizarse cada día que se use el esterilizador, antes de la primera carga procesada, en esterilizadores de remoción dinámica de aire.
Integridad del sistema: fugas, alarmas, interlocks y registradores
La OQ también cubre pruebas de integridad y seguridad: alarmas, bloqueos (interlocks), válvulas, pruebas de fuga y la consistencia del registro impreso o digital.
Esto importa porque, si el registro del ciclo es incompleto o puede alterarse, la trazabilidad se debilita.
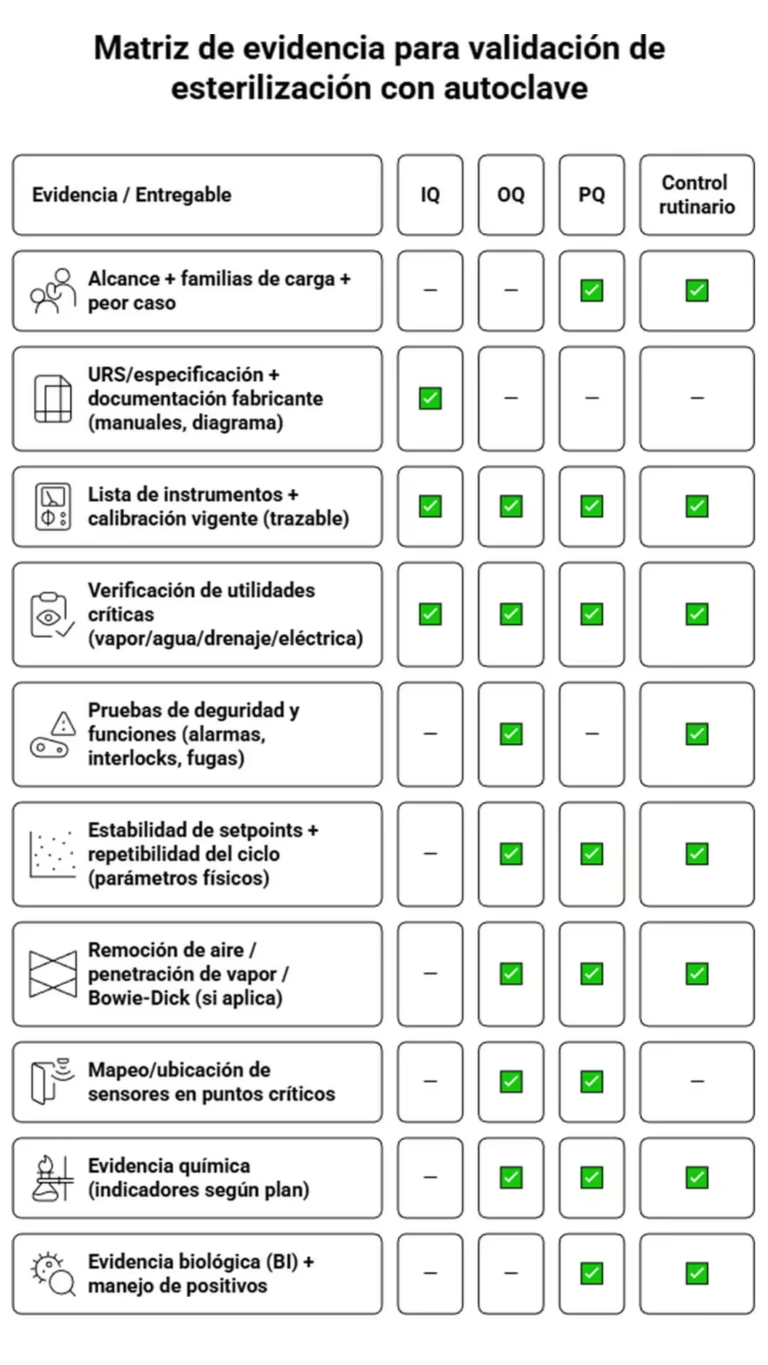
PQ: Calificación de desempeño en cargas reales
La PQ responde a lo que realmente importa: ¿la esterilización con autoclave funciona en condiciones reales con cargas representativas y con el peor caso?
Cómo se realiza la validación del proceso de esterilización en la práctica
La PQ se construye con:
- Definición y justificación de cargas (familias y peor caso),
- Corridas repetidas bajo condiciones definidas,
- Monitoreo físico con sensores en puntos críticos,
- Uso de indicadores químicos y biológicos según el plan,
- Evaluación de secado y condición del empaque,
- Análisis, desviaciones, conclusiones y criterios de liberación.
En hospitales, el desafío no es “hacer muchas corridas”, sino que cada corrida responda a una pregunta clara y produzca evidencia utilizable.
Indicadores biológicos: qué miden y por qué se usan (Geobacillus stearothermophilus)
El Centro para el Control y Prevención de Enfermedades (CDC) indica que la efectividad de la esterilización por vapor se monitorea con un indicador biológico.
Este contiene esporas de Geobacillus stearothermophilus y los casos positivos, aunque raros, pueden atribuirse a error operativo, entrega de vapor inadecuada o fallas del equipo.
En cuanto al estándar del insumo, ISO 11138-3 especifica requisitos para indicadores biológicos destinados a procesos de esterilización por calor húmedo (vapor).
La práctica recomendada es tratar el indicador biológico como un instrumento:
- Controlar lote
- Condiciones de almacenamiento
- Controles positivos/negativos según procedimiento
- Trazabilidad del resultado.
Paquetes de desafío y ubicación de sensores
Para que la PQ tenga fuerza, los sensores se ubican donde es más probable que exista “punto frío” o peor penetración:
- Dentro de contenedores rígidos
- Centros de paquetes porosos densos
- Zonas internas del set más desafiante.
Esta lógica se alinea con el enfoque de EN 285 sobre penetración y causas de fallos relacionadas con aire residual o gases no condensables.
Control rutinario: cómo se puede verificar un proceso de esterilización cada día
Una vez validado, el proceso requiere verificación rutinaria para confirmar que se mantiene “dentro de lo validado”.
Esto responde directamente a la pregunta: ¿cómo se puede verificar un proceso de esterilización?
Controles por jornada y por carga: qué no debe faltar
En términos de mínimos operativos:
- Revisión de parámetros físicos del ciclo en cada carga (tiempo/temperatura/presión según ciclo),
- Prueba Bowie-Dick diaria en pre-vacío antes de la primera carga (cuando aplique),
- Uso de indicadores químicos conforme a política (al menos indicador de proceso externo en paquetes),
- Empleo de indicadores biológicos según programa y riesgo.
El CDC refuerza el papel del indicador biológico para monitoreo de vapor y la interpretación de positivos como eventos que requieren investigación.
La zona limpia es donde se transforma “un dispositivo limpio” en “un dispositivo listo para esterilizarse y mantenerse estéril hasta el punto de uso”.

Indicadores químicos (ISO 11140): tipo 1, tipo 2, integradores y su interpretación
ISO 11140-1 define requisitos para indicadores químicos usados en procesos de esterilización. En la práctica, los programas suelen distinguir:
- Tipo 1 (indicador de proceso): evidencia exposición.
- Tipo 2 (uso específico): como Bowie-Dick para remoción de aire, regulado por ISO 11140-5.
- Integradores (frecuentemente tipo 5): diseñados para responder a variables críticas del proceso.
Como referencia didáctica, fabricantes y centros técnicos suelen explicar la clasificación de “tipos” basada en ISO 11140-1 (por ejemplo, el esquema 1–6).
El punto clave es interpretar correctamente: un indicador químico no sustituye la evidencia física del ciclo, ni reemplaza un indicador biológico cuando la política/riesgo lo exige.
Cada uno cumple una función distinta dentro de la cadena de evidencia.
Registros, liberación de carga y trazabilidad clínica
Un sistema robusto documenta quién cargó, qué se cargó, qué ciclo se ejecutó, qué resultados de monitoreo se obtuvieron y quién liberó la carga.
La trazabilidad es coherente con la orientación de buenas prácticas en reprocesamiento de la OMS.
Estas describen la secuencia completa desde recepción hasta almacenamiento / distribución.
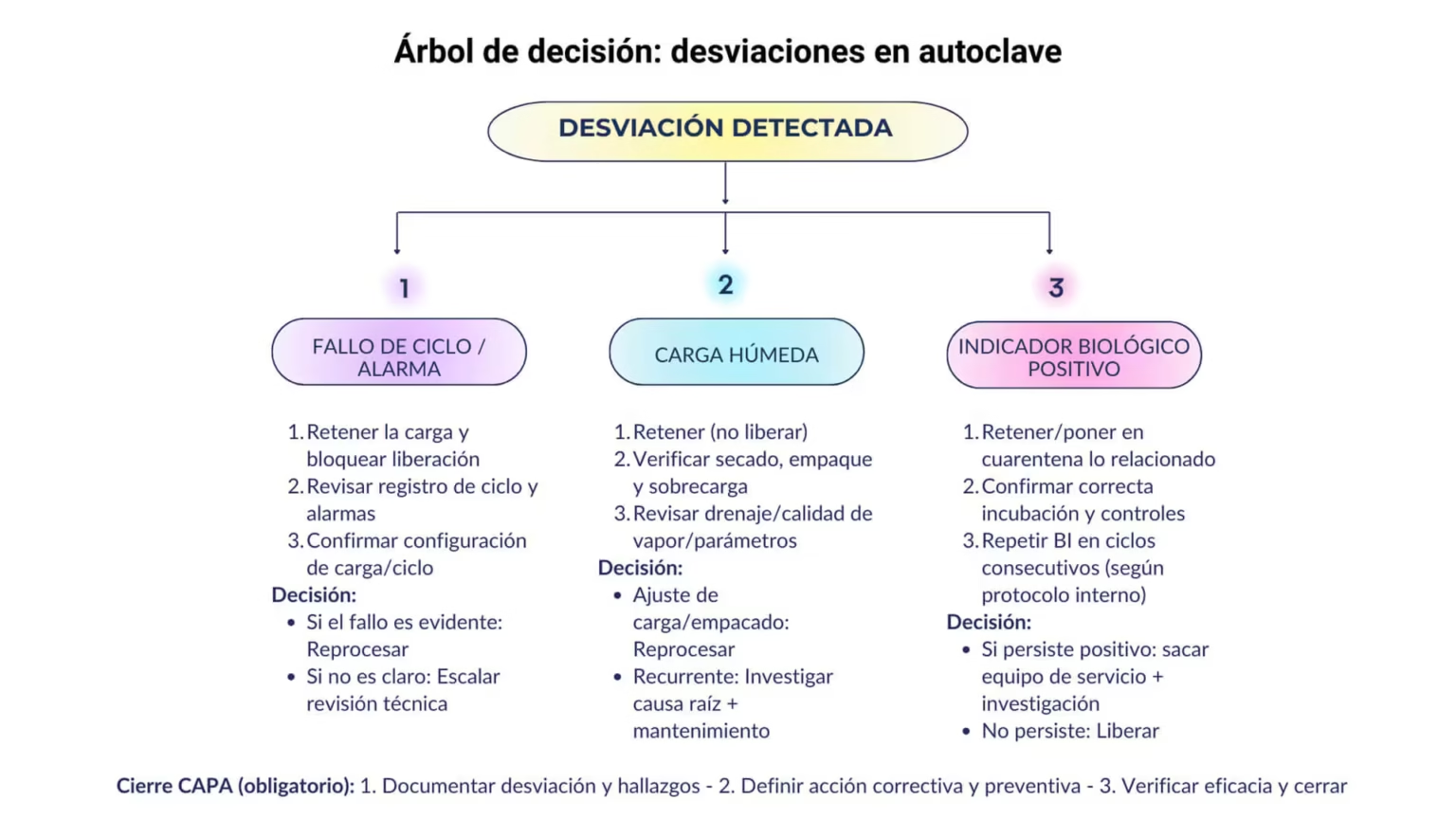
Qué hacer ante desviaciones: fallos de ciclo, cargas húmedas y biológico positivo
Una central con cultura de validación entiende que el error ocurre, pero no se “barre bajo la alfombra”. Se investiga, se corrige y se documenta.
Investigación de causa raíz y retención de material
Ante un fallo de ciclo o carga húmeda, la respuesta típica incluye:
- Retener la carga (no liberar),
- Revisar registro físico del ciclo,
- Verificar configuración de carga y empaque,
- Evaluar condiciones de vapor y drenajes,
- Revisar mantenimiento reciente y alarmas,
- Documentar hallazgos y acciones.
EN 285 y guías técnicas coinciden en que problemas de remoción de aire, fugas o gases no condensables pueden impactar la penetración del vapor y el resultado del proceso.
Protocolo sugerido ante indicador biológico positivo (vapor)
El CDC publica un protocolo sugerido para el manejo de un indicador biológico positivo. Entre los puntos clave están:
- Repetir el indicador biológico en tres ciclos consecutivos lo antes posible
- Considerar retiro/recall de material procesado desde el último resultado aceptable si persisten positivos
- Evaluar si el esterilizador o el procedimiento están defectuosos.
Esto suele acompañarse de revisión inmediata de pruebas funcionales (por ejemplo, Bowie-Dick si aplica) y revisión de carga/ciclo.
Adicionalmente, la confirmación de que el indicador biológico fue manipulado e incubado correctamente, con su control positivo.
Cómo elegir un equipo de validación de esterilización para autoclaves en clínicas y hospitales
La mejor respuesta a esta pregunta, muy frecuente, es elegirlo por su capacidad de producir evidencia defendible.
Data loggers, sensores y calibración trazable (ISO/IEC 17025, ONAC)
Los registradores de temperatura/presión y sensores deben tener calibración confiable.
La norma ISO/IEC 17025 es el estándar internacional para laboratorios de ensayo y calibración.
Define los requisitos de competencia, imparcialidad y operación consistente, precisamente para asegurar resultados confiables.
En Colombia, ONAC mantiene el Directorio Oficial de Acreditados para consultar organismos acreditados (incluyendo laboratorios de calibración ISO/IEC 17025).
En términos prácticos, al seleccionar el equipo de validación, conviene exigir:
- Calibración vigente y trazable,
- Especificaciones de exactitud e incertidumbre,
- Estabilidad a temperatura y humedad del entorno,
- Software que proteja integridad del dato (usuario, auditoría, exportación).
Software de validación y gestión documental
El software suma valor cuando ayuda a controlar versiones, firmas, auditoría de cambios y consolidación de reportes.
No es un lujo: en validación, perder un dato o no poder demostrar integridad documental reduce la fuerza del informe.
Productos recomendados para la validación de autoclaves en hospitales
La pregunta no debería resolverse con “marcas”, sino con “tipos” de productos y su función en el sistema de evidencia.
Qué insumos elegir según riesgo y tipo de autoclave
Una guía práctica para elegir insumos considera:
- Tipo de esterilizador (gravedad vs pre-vacío),
- Familias de carga (porosa, metálica, contenedor, lúmenes),
- Criticidad clínica (implantes vs no implantes),
- Frecuencia de uso y nivel de control requerido.
En esterilizadores de pre-vacío, un insumo típico es el paquete Bowie-Dick (tipo 2) bajo ISO 11140-5 para prueba de remoción de aire.
Consumibles críticos: Bowie-Dick, integradores y biológicos
Un conjunto razonable, sin caer en exceso, suele incluir:
- Indicadores de proceso (tipo 1) para paquetes/cargas, alineados a ISO 11140-1.
- Bowie-Dick (tipo 2) en pre-vacío, según ISO 11140-5.
- Indicadores biológicos para vapor conformes con ISO 11138-3, con microorganismo objetivo (G. stearothermophilus según CDC para monitoreo de vapor).
Cuando la institución maneja “cargas peor caso” complejas, también se usan dispositivos de desafío del proceso que simulan la mayor dificultad de penetración.
Qué empresas ofrecen servicios de validación de esterilización en autoclaves en Colombia
Esta pregunta es válida, pero la respuesta técnica responsable empieza aclarando algo: no hay un listado oficial único que cubra todo el mercado.
Por eso, lo más seguro es evaluar proveedores por criterios verificables y por entregables.
Criterios técnicos para evaluar proveedores y entregables
Al solicitar propuestas de servicios de validación (IQ/OQ/PQ) en Colombia, conviene exigir:
- Protocolo alineado a ISO 17665 e ISO/TS 17665-2 (alcance, criterios de aceptación, peor caso).
- Trazabilidad metrológica: instrumentos calibrados y evidencia de competencia de calibración (ISO/IEC 17025), verificable en ONAC cuando aplique.
- Entregables auditables: datos crudos, análisis, desviaciones, conclusiones, recomendaciones y plan de control rutinario.
- Manejo de desviaciones: criterio claro ante biológico positivo, alineado con guías como CDC.
- Experiencia demostrable en centrales hospitalarias, porque las cargas mixtas y la operatividad hospitalaria tienen desafíos propios.
Un criterio adicional, cuando la institución opera con alta demanda, es revisar si el proveedor sabe diseñar validaciones para escenarios con dos autoclaves de esterilización (redundancia operativa), con protocolos que diferencien evidencia por equipo, ciclo y familias de carga.
Una validación robusta de esterilización con autoclave se reconoce porque no depende de “confianza”, sino de evidencia.
Debe ser un proceso definido, IQ/OQ/PQ bien ejecutados, control rutinario proporcional al riesgo, y gestión de desviaciones con trazabilidad.
Ese enfoque protege al paciente, reduce reprocesos y fortalece auditorías, sin necesidad de complejidad innecesaria.
Si tu institución está ajustando su programa de validación o necesita estructurar protocolos IQ/OQ/PQ y el plan de control rutinario con respaldo documental, el acompañamiento de un equipo técnico con experiencia en centrales hospitalarias puede acelerar el cierre de brechas y dejar evidencia lista para auditoría.
Preguntas frecuentes (FAQ)
Es la demostración documentada de que el autoclave y el proceso definido esterilizan de manera consistente cargas definidas, con criterios de aceptación y control rutinario. ISO 17665 estructura esa exigencia como desarrollo, validación y control rutinario del proceso de calor húmedo.
El método más utilizado es IQ/OQ/PQ: instalación (IQ), operación (OQ) y desempeño con cargas reales (PQ), apoyado en monitoreo físico, indicadores químicos e indicadores biológicos según programa y riesgo.
Se define el proceso y las familias de carga, se ejecuta IQ (instalación/documentación), OQ (pruebas funcionales, remoción de aire, estabilidad) y PQ (desempeño con cargas peor caso), con reportes auditables y plan de control rutinario.
Con control rutinario: revisión de parámetros físicos de cada ciclo, indicadores químicos adecuados al proceso y, según el programa, indicadores biológicos. Para vapor, el CDC señala que el monitoreo de efectividad se hace con indicadores biológicos de G. stearothermophilus y define acciones ante positivos.
En práctica hospitalaria/farmacéutica se usa el esquema de tres calificaciones: IQ, OQ y PQ, coherente con el ciclo de vida de validación y control descrito en ISO 17665.
Primero, cargar y empacar de forma estandarizada, evitando configuraciones improvisadas que cambien la penetración del vapor. Segundo, ejecutar y documentar controles rutinarios (incluida Bowie-Dick cuando aplique en pre-vacío). Tercero, tratar desviaciones con investigación y evidencia, especialmente ante biológico positivo, siguiendo protocolos sugeridos como los del CDC.
Se recomienda alinear el programa con la Resolución 2183 de 2004 (Manual de Buenas Prácticas de Esterilización) y con la Resolución 914 de 2025 (Manual DMER), manteniendo registros y evidencia compatibles con exigencias de habilitación (Resolución 3100 de 2019). Para estructurar el “cómo” técnico de la evidencia, ISO 17665 e ISO/TS 17665-2 son referencias sólidas para validación y control rutinario.Principio del formularioFinal del formulario



